Tilbake til artikkelserien om metabolismen
Medfødte metabolske sykdommer kan ramme nesten hele metabolismen. Selv om disse sykdommene er veldig sjeldne, så er konsekvensene store for de som får dem. I denne artikkelen får du en oversikt over en del av de sykdommene som rammer omsetningen av karbohydrater.
Relevante artikler:
- Karbohydrater
- Glukosemetabolismen
- Glykogenmetabolismen
- Fruktose- og galaktosemetabolismen
- Genetikk
- Metabolske sykdommer – prinsippene
Jeg vil også anbefale en artikkel av Mayatepek og medarbeidere (1), som gir en god innføring i de ulike sykdommmene.
Sykdommer i glykogenmetabolismen
Glykogen er lagringsformen for karbohydrater, og spesielt leverglykogenet er veldig sentralt i reguleringen av blodsukkeret. Når dette fungerer som det skal kan leveren regulere blodsukkeret med god presisjon ved å lagre inn glukoseoverskudd og slippe ut glukose når blodsukkeret synker. Dette er imidlertid avhengig av en rekke enzymer, og medfødte defekter i disse kan ha negative effekter som først og fremst rammer reguleringen av blodsukkeret og dermed hele kroppens tilførsel av glukose. En vanlig konsekvens er hepatomegali, altså unormal vekst av leveren. Hyperlipidemi kan også oppstå som følge av at leveren må produsere mer fettsyrer enn vanlig. Glykogenlagringssykdom (GLS) er et paraplybegrep for flere sykdommer som rammer glykogenmetabolismen, og disse rammer omtrent 1 av 20-40 000. Her vil jeg bare beskrive de tre viktigste variantene.
GLS 1 er den klassiske varianten, hvor man har en dysfunksjon i enzymet glukose-6-fosfatase. Dette enzymet defosforylerer glukose-6-fosfat slik at det dannes fri glukose som kan krysse cellemembranen og dermed havne i blodet. Uten dette enzymet vil ikke leveren (eller nyrene) være i stand til å frigi glukose, noe som medfører at man blir avhengig av kontinuerlig tilførsel av glukose fra kostholdet. Korte fasteperioder kan medføre hypoglykemi.
GLS 3 skyldes en defekt i enzymet som spalter forgreningene i glykogenmolekylet. Dette medfører at kapasiteten for å frigjøre glukose fra glykogenet er kraftig redusert. Her vil imidlertid glukoneogenesen fungere som normalt, slik at risikoen for hypoglykemi ved faste er mye lavere enn ved GLS 1.
GLS 6 og 9 rammer glykogen fosforylase, som er enzymet som spalter av glukosemolekyler fra glykogenet. Også her vil glukoneogenesen kunne gå som normalt, og hypoglykemi er ikke normalt en komplikasjon. Ettersom alkohol hemmer glukoneogenesen bør personer med disse sykdommene være forsiktig med inntaket av dette.
Sykdommer i fruktosemetabolismen
Fruktose er et av de tre monosakkaridene. Vi finner fruktose som en del av helt vanlig hvitt sukker, siden fruktosen utgjør 50 % av sukrosemolekylet. I tillegg til dette finner vi fri fruktose i frukt og noe i grønnsaker. Vi kjenner til tre ulike enzymdefekter i fruktosemetabolismen, som har ulike konsekvenser.
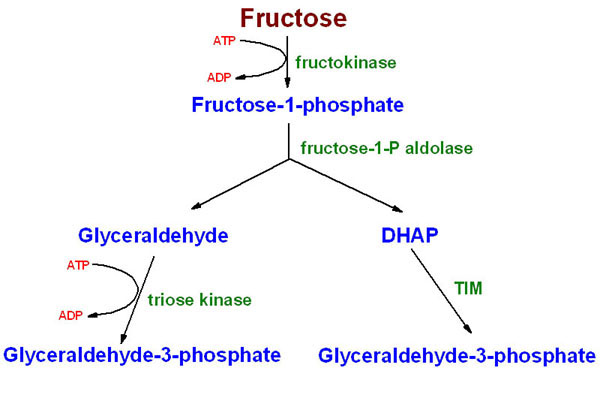
Essensiell fruktosuri skyldes mangel på eller defekt i enzymet fruktokinase, som fosforylerer fruktose og danner fruktose-1-fosfat (F1P). Dette er en ufarlig tilstand som rammer omtrent 1 av 130 000, og konsekvensene av dette er at vi skiller ut fruktose i urinen.
Arvelig fruktoseintoleranse er en alvorlig defekt som skyldes mangel på Aldolase B, som metaboliserer fruktose-1-fosfat. Dette rammer omtrent 1 av 20 000. Mangel på denne aldolasen fører til at F1P hoper seg opp i leveren. Opphopning av F1P fører til at inorganisk fosfat bindes opp her, slik at levercellene ikke kan produsere nok ATP, og dette går utover leverens funksjoner. Blant konsekvensene er at både nedbrytning av glykogen og leverens glukoneogenese hemmes, noe som medfører hypoglykemi. Også proteinsyntesen i leveren er energikrevende, og mangel på ATP vil ramme denne. Dette går blant annet utover produksjonen av viktige plasmaproteiner og koagulasjonsfaktorer. Arvelig fruktoseintoleranse behandles med et strengt, fruktosefritt kosthold, noe som er en utfordring og gjør det vanskelig å dekke alle behov for næringsstoffer.
Den siste defekten i fruktosemetabolismen er mangel på fruktose-1,6-bisfosfatase, som er et sentralt enzym i glukoneogenesen. Forekomsten av dette er ikke godt kartlagt, men den kan ligge rundt 1 av 350 000. Etter hvert som leverglykogenet begynner å tømmes er vi avhengig av glukoneogenesen for å produsere glukose til blodsukkeret. Mangel på dette enzymet kan derfor ha alvorlige konsekvenser, ettersom det medfører at glukoneogenesen ikke virker. I tillegg vil det medføre økt produksjon av ketonlegemer og andre sure metabolitter, slik at vi også utvikler en acidose. Behandlingen av fruktose-1,6-bisfosfatasemangel sikter mot å fylle glykogenlagrene og redusere behovet for glukoneogenese. Dette oppnås først og fremst ved å unngå lengre fasteperioder, samtidig som karbohydrater og da spesielt stivelse, må utgjøre en solid del av kostholdet.
Sykdommer i galaktosemetabolismen
Galaktose er et monosakkarid som hovedsakelig finnes i mat som en bestanddel av disakkaridet laktose (melkesukker). Det finnes litt fri galaktose i frukt og grønnsaker, og i tillegg produserer vi også litt selv hver dag fra glukose. Galaktose er essensielt for oss, blant annet som en del av en rekke glykoproteiner/glykolipider. Fra artikkelen om galaktosemetabolismen husker vi at vi har tre viktige enzymer som sørger for omdanning til glukose: galaktokinase (GALK), galaktose-1-fosfat uridyltransferase (GALT) og UDP-galaktose-4-epimerase (GALE). Alle disse kan rammes, og vi har derfor tre kjente sykdommer i galaktosemetabolismen. Disse kalles galaktosemi som følge av at galaktosekonsentrasjonen i blodet øker.
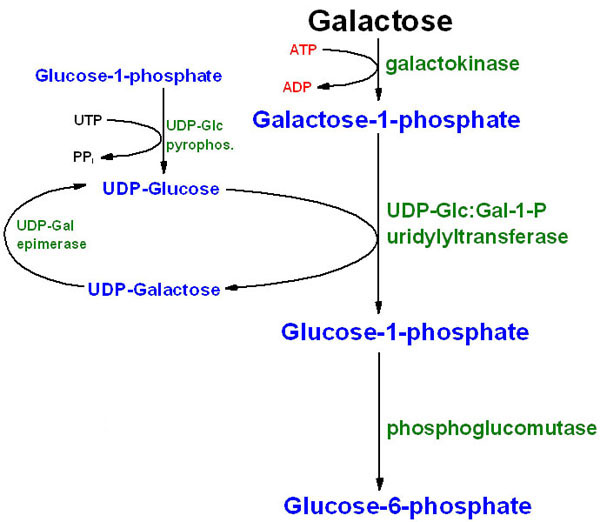
Klassisk galaktosemi skyldes en defekt i GALT, og prevalensen er 1 av 47 000. Denne defekten medfører en opphopning av galaktose-1-fosfat (og galaktose), som da må metaboliseres på andre måter. Galaktose-1-fosfat metaboliseres da til to forbindelser, galaktitol og galaktonat, som også hoper seg opp. Galaktitol kan blant annet hope seg opp i øyet og forårsake grå stær, mens galaktonat er antatt å kunne medføre akutt forgiftning av lever og nyrer.
Klassisk galaktosemi er den mest alvorlige defekten i galaktosemetabolismen, men også defekter i de to andre enzymene finnes. Dersom GALK ikke fungerer vil ikke galaktosen fosforyleres. Galaktose kan metaboliseres og danne galaktitol, med grå stær som konsekvens, men mesteparten av galaktosen vil skilles ut uendret. Ved defekter i GALE vil vi ikke være i stand til å metabolisere UDP-galaktose til UDP-glukose, men heller ikke den motsatte reaksjonen kan skje. Vi får derfor heller ikke produsert UDP-galaktose fra glukose, noe som kan medføre mangel på substrat for å danne glykosylerte forbindelser. Defekter i GALE er ekstremt sjelden, men her kan det være nødvendig å finberegne inntaket av galaktose for å gi nok til å danne glykosylerte forbindelser men ikke så mye at vi får opphopninger av metabolitter lenger oppe i galaktosemetabolismen.
Referanser:
- Mayatepek E, Hoffmann B, Meissner T: Inborn errors of carbohydrate metabolism. Best practice & research Clinical gastroenterology 2010, 24(5):607-618.